





Além da tecnologia, a síntese de glicosídeos sempre despertou interesse na ciência, por ser uma reação muito comum na natureza. Artigos recentes de Schmidt, Toshima e Tatsuta, bem como muitas referências neles citadas, comentaram sobre uma ampla gama de potenciais sintéticos.
Na síntese de glicosídeos, um componente multiaçúcar é combinado com nucleófilos, como álcoois, carboidratos ou proteínas. Se uma reação seletiva com um dos grupos hidroxila de um carboidrato for necessária, todas as outras funções devem ser protegidas na primeira etapa. Em princípio, processos enzimáticos ou microbianos, devido à sua seletividade, podem substituir etapas complexas de proteção e desproteção química para seletivamente remover glicosídeos de regiões específicas. No entanto, devido à longa história dos alquil glicosídeos, a aplicação de enzimas na síntese de glicosídeos ainda não foi amplamente estudada e aplicada.
Devido à capacidade de sistemas enzimáticos adequados e aos altos custos de produção, a síntese enzimática de alquil poliglicosídeos não está pronta para ser atualizada para o nível industrial, sendo preferidos métodos químicos.
Em 1870, MAcolley relatou a síntese de “acetocloridrose” (1, figura 2) pela reação de dextrose (glicose) com cloreto de acetila, o que eventualmente levou à história das rotas de síntese de glicosídeos.
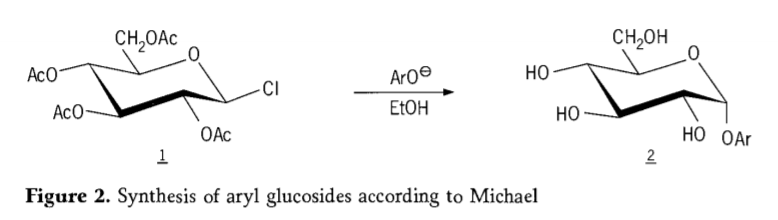
Halogenetos de tetra-0-acetil-glicopiranosila (acetohaloglicoses) foram posteriormente descobertos como intermediários úteis para a síntese estereosseletiva de alquil glicosídeos puros. Em 1879, Arthur Michael conseguiu preparar aril glicosídeos cristalizáveis definidos a partir de intermediários de Colley e fenolatos. (Aro-,Figura 2).
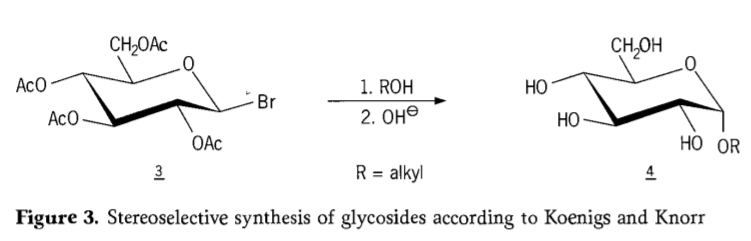
Em 1901, Michael desenvolveu a síntese de uma ampla gama de carboidratos e agliconas hidroxílicas, quando W. Koenigs e E. Knorr introduziram seu processo aprimorado de glicosidação estereosseletiva (Figura 3). A reação envolve uma substituição de SN2 no carbono anomérico e prossegue estereosseletivamente com inversão de configuração, produzindo, por exemplo, o α-glicosídeo 4 a partir do anômero β do intermediário aceobromoglicose 3. A síntese de Koenigs-Knorr ocorre na presença de promotores de prata ou mercúrio.
Em 1893, Emil Fischer propôs uma abordagem fundamentalmente diferente para a síntese de alquil glicosídeos. Esse processo é hoje conhecido como "glicosidação de Fischer" e compreende uma reação catalisada por ácido de glicoses com álcoois. Qualquer relato histórico, no entanto, deve incluir também a primeira tentativa relatada por A. Gautier, em 1874, de converter dextrose com etanol anidro na presença de ácido clorídrico. Devido a uma análise elementar enganosa, Gautier acreditou ter obtido uma "diglicose". Fischer posteriormente demonstrou que a "diglicose" de Gautier era, na verdade, principalmente etil glicosídeo (Figura 4).
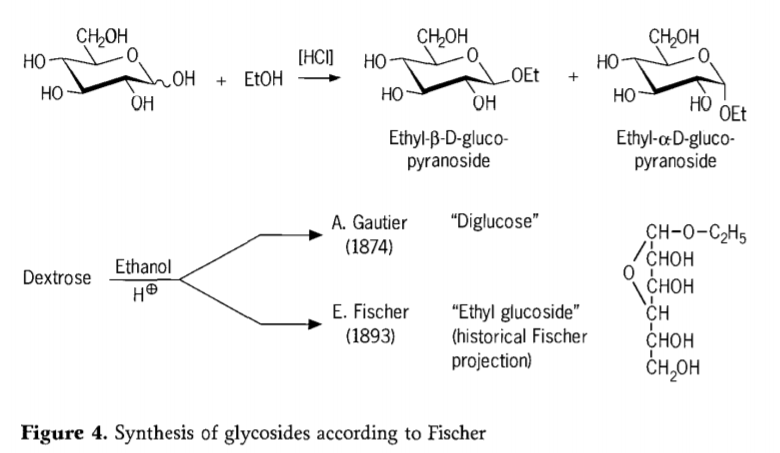
Fischer definiu corretamente a estrutura do etil glicosídeo, como pode ser observado na fórmula furanosídica histórica proposta. De fato, os produtos de glicosidação de Fischer são misturas complexas, em sua maioria em equilíbrio, de anômeros α/β e isômeros de piranosídeo/furanosídeo, que também compreendem oligômeros de glicosídeos ligados aleatoriamente.
Consequentemente, espécies moleculares individuais não são fáceis de isolar das misturas de reação de Fischer, o que representou um sério problema no passado. Após algum aprimoramento desse método de síntese, Fischer posteriormente adotou a síntese de Koenigs-Knorr para suas investigações. Utilizando esse processo, E. Fischer e B. Helferich foram os primeiros a relatar a síntese de um alquil glicosídeo de cadeia longa com propriedades surfactantes em 1911.
Já em 1893, Fischer havia corretamente observado propriedades essenciais dos alquil glicosídeos, como sua alta estabilidade à oxidação e hidrólise, especialmente em meios fortemente alcalinos. Ambas as características são valiosas para alquil poliglicosídeos em aplicações surfactantes.
Pesquisas relacionadas à reação de glicosidação ainda estão em andamento e diversas rotas interessantes para a obtenção de glicosídeos foram desenvolvidas recentemente. Alguns dos procedimentos para a síntese de glicosídeos estão resumidos na Figura 5.
Em geral, os processos de glicosidação química podem ser divididos em processos que levam a equilíbrios complexos de oligômeros na troca de glicosil catalisada por ácido.
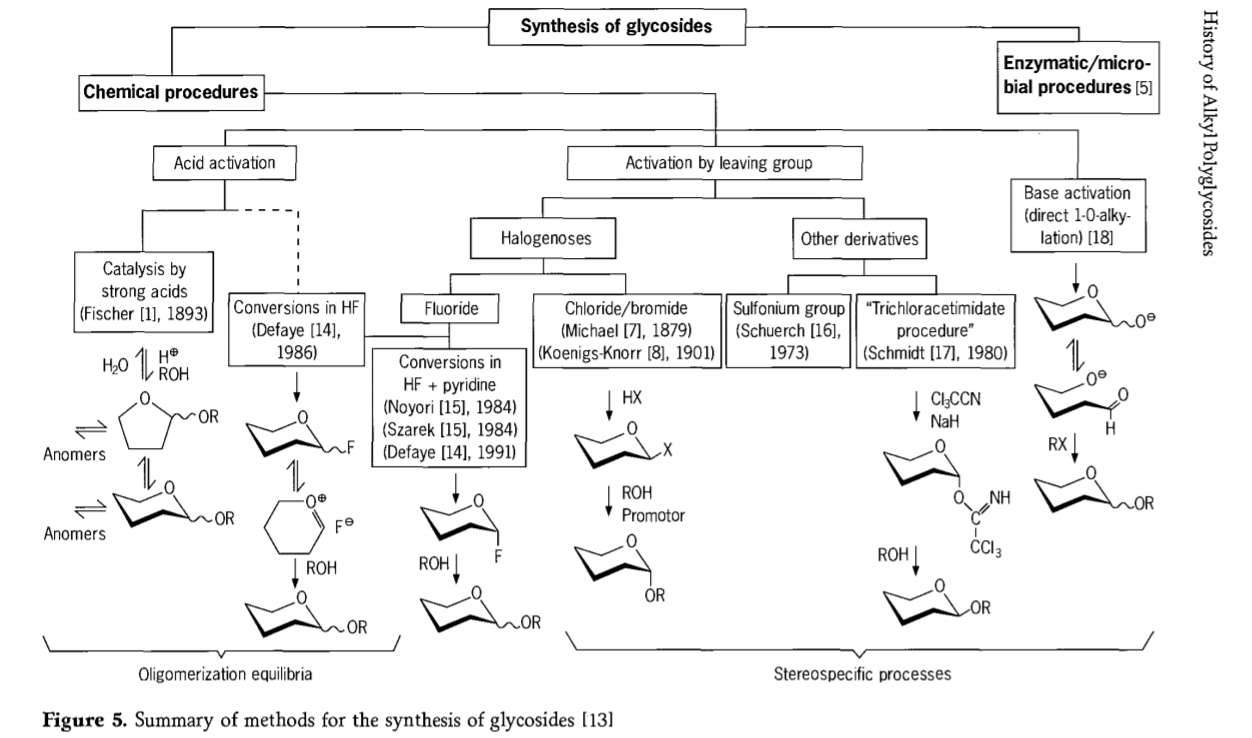
Reações em substratos de carboidratos apropriadamente ativados (reações glicosídicas de Fischer e reações de fluoreto de hidrogênio (HF) com moléculas de carboidratos desprotegidas) e reações de substituição cinética controlada, irreversíveis e principalmente estereotáxicas. Um segundo tipo de procedimento pode levar à formação de espécies individuais em vez de misturas complexas de reações, especialmente quando combinado com técnicas de grupos de conservação. Carboidratos podem deixar grupos no carbono ectópico, como átomos de halogênio, sulfonilas ou grupos tricloroacetimidato, ou ser ativados por bases antes da conversão em ésteres triflato.
No caso particular de glicosidações em fluoreto de hidrogênio ou em misturas de fluoreto de hidrogênio e piridina (poli(fluoreto de hidrogênio) de piridínio), glicosil fluoretos são formados in situ e são suavemente convertidos em glicosídeos, por exemplo, com álcoois. O fluoreto de hidrogênio demonstrou ser um meio de reação fortemente ativador e não degradante; a autocondensação em equilíbrio (oligomerização) é observada de forma semelhante ao processo de Fischer, embora o mecanismo de reação seja provavelmente diferente.
Alquiglicosídeos quimicamente puros são adequados apenas para aplicações muito especiais. Por exemplo, alquilglicosídeos têm sido utilizados com sucesso em pesquisas bioquímicas para a cristalização de proteínas de membrana, como a cristalização tridimensional de porina e bacteriorrodopsina na presença de octil β-D-glicopiranosídeo (experimentos posteriores baseados neste trabalho levaram Deisenhofer, Huber e Michel ao Prêmio Nobel de Química em 1988).
Durante o desenvolvimento de alquil poliglicosídeos, métodos estereosseletivos têm sido utilizados em escala laboratorial para sintetizar uma variedade de substâncias modelo e estudar suas propriedades físico-químicas. Devido à sua complexidade, à instabilidade dos intermediários e à quantidade e natureza crítica dos resíduos do processo, sínteses do tipo Koenigs-Knorr e outras técnicas de grupos protetores criariam problemas técnicos e econômicos significativos. Os processos do tipo Fischer são comparativamente menos complicados e mais fáceis de realizar em escala comercial e, portanto, são o método preferido para a produção de alquil poliglicosídeos em larga escala.
Data de publicação: 12 de setembro de 2020